
The Invisible Interfaces That Shape Chromatographic Truth
Why every wetted LC interface, from sample handling to detection, must be treated as part of the analytical method


In liquid chromatography (LC), the surfaces encountered during sample aspiration, storage, injection, separation, and detection can determine whether a method produces accurate, reproducible data or introduces systematic bias. These interfaces contribute to nonspecific adsorption, analyte loss, peak tailing, carryover, metal adduct formation, and ion suppression, sometimes even after extensive column and mobile-phase optimization. Surface chemistry must therefore be treated as a primary method-development variable, not as a secondary troubleshooting concern. The analytical target profile is incomplete unless it considers the analyte's surface-interaction risk, the workflow, and the separation mode. This article proposes a practical Class I–IV framework for assessing surface-interaction risk and aligning consumables, hardware, mobile-phase design, robustness studies, and lifecycle controls with the expected analyte behavior. The discussion draws on peer-reviewed literature [1], [2], [4] and connects these considerations to ICH Q2(R2) [6], ICH Q14 [7], and USP <1220> [8].
The Hidden Variable in Chromatographic Performance
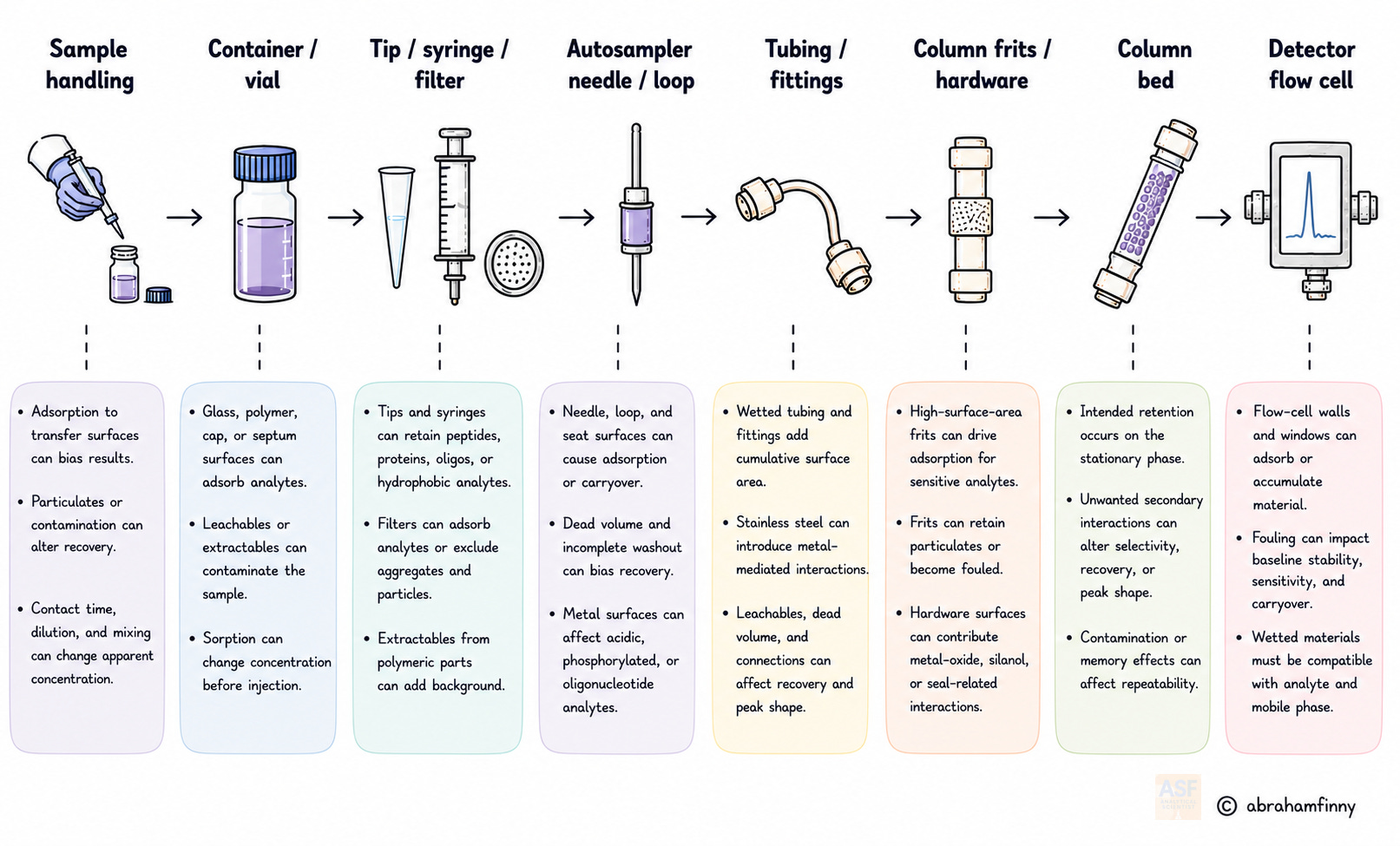
Chromatographers routinely invest substantial effort in stationary-phase chemistry, mobile-phase optimization, and gradient design. Yet the non-column surfaces encountered by the sample, including pipette tips, vial walls, tubing, frits, injection needles, and detector components, often receive far less systematic attention. At trace levels, even modest adsorption can cause measurable losses, compromised limits of quantification, and method failures that only appear after validation, transfer, or routine use.
Nonspecific adsorption results from the same intermolecular forces involved in deliberate chromatographic retention: electrostatic interactions, hydrophobic partitioning, hydrogen bonding, and Lewis acid–base coordination. However, these forces act on uncontrolled surfaces shaped by manufacturing rather than analytical design. The cumulative wetted surface area encountered during sample handling, injection, and flow through high-surface-area components such as frits can amplify small partitioning effects into measurable bias. Understanding these interfaces at the molecular level is therefore essential for developing methods that remain reliable across validation, transfer, and routine operation.
Surface chemistry is therefore a lifecycle variable, not merely a troubleshooting variable. ICH Q2(R2) [6] and USP <1220> [8] require evidence that an analytical procedure is fit for its intended purpose. Yet surface-contact points are not always explicitly challenged during development, validation, transfer, or routine control. For high-risk analytes, especially oligonucleotides, phosphopeptides, proteins, and gene-therapy materials, a method can satisfy conventional system-suitability criteria while still carrying hidden recovery or transfer risk if consumables and hardware surfaces have not been evaluated.
Fundamental Mechanisms of Surface–Analyte Interactions
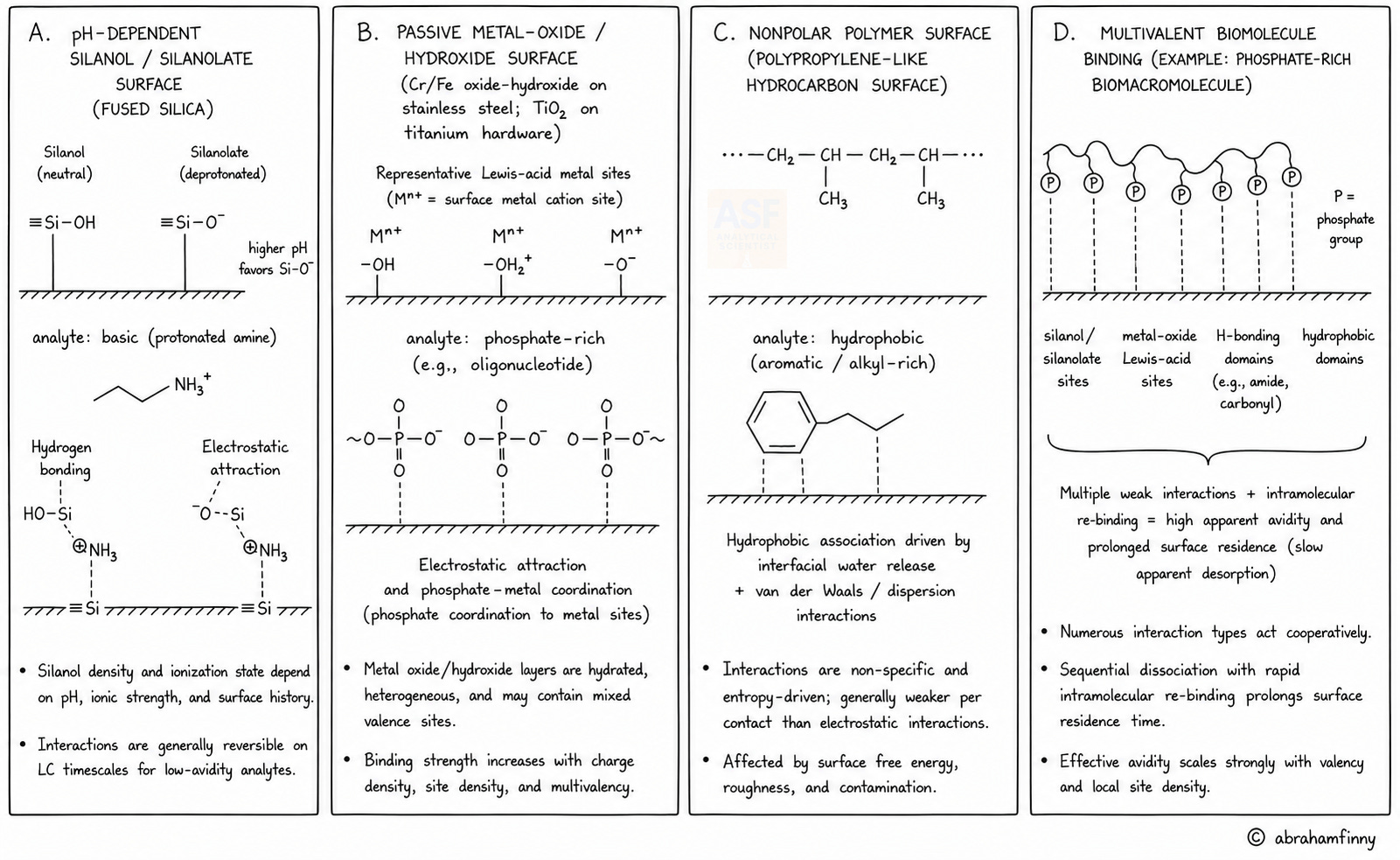
Surface adsorption in chromatography follows established thermodynamic and kinetic principles. The free energy of adsorption reflects both enthalpic contributions, including electrostatic attraction, metal coordination, hydrogen bonding, and dispersion forces, and entropic contributions, including the release of ordered water at hydrophobic interfaces. At low surface coverage, adsorption may approximate Langmuirian behavior; on heterogeneous or partially conditioned surfaces, Freundlich-type behavior is often more realistic. Residence time and flow-path geometry also matter: stagnant zones, frit porosity, dead volumes, and repeated aspirate–dispense cycles increase the opportunity for unintended retention.
The key distinction is not simply whether adsorption occurs, but whether desorption remains fast enough on the chromatographic timescale. Small molecules often interact through a single dominant functional group; when adsorption occurs, it can be reduced by pH adjustment, ionic-strength optimization, selection of organic modifiers, or the addition of competing additives. Biomolecules and advanced therapeutic modalities exhibit distinct adsorption regimes. Oligonucleotides, phosphopeptides, proteins, conjugates, viral vectors, extracellular vesicles, plasmid DNA, and mRNA-LNP systems can make multiple simultaneous contacts with the same surface. This multivalency can convert individually weak interactions into persistent adsorption, slow recovery, conditioning effects, and apparent irreversibility during routine LC analysis.
At the molecular level, different surfaces exhibit distinct binding motifs. Glass and fused silica expose silanol and siloxane domains, respectively. Stainless steel and titanium exhibit passive oxide or hydroxide layers that contain Lewis-acidic metal centers. Polymeric consumables introduce hydrophobic adsorption, wetting, and extractables-related risks. The practical consequence is that surface adsorption is rarely governed by a single mechanism. A basic small molecule may be dominated by silanol exchange; a phosphorylated peptide may be dominated by metal coordination; a protein or oligonucleotide may combine charge, coordination, hydrogen bonding, hydrophobic contact, and conformational adaptation. This mechanistic diversity is why surface chemistry must be evaluated across the actual workflow rather than inferred only from the analytical column. The major surface classes in LC workflows should therefore be treated as distinct chemical environments, each with a characteristic adsorption profile.
Glass and Fused-Silica Surfaces
Borosilicate glass (Type I) presents a heterogeneous oxide surface containing silanol (Si–OH) groups and siloxane (Si–O–Si) bridges. Depending on surface hydration, local composition, pH, ionic strength, and manufacturing history, silanol groups can deprotonate, forming negatively charged sites that interact electrostatically with protonated basic analytes. Siloxane-rich regions and adsorbed organic residues can also contribute to hydrophobic adsorption, particularly for lipophilic or amphiphilic compounds. This mixed ionic and hydrophobic character helps explain the variable recovery sometimes observed for basic or surface-active analytes in untreated glass vials and is mechanistically related, though not identical, to residual silanol effects observed in silica-based LC columns [17], [18].
Silanization with monofunctional or difunctional chlorosilanes caps reactive Si–OH groups, converting the surface to a more hydrophobic trimethylsilyl or longer-chain layer. While effective at suppressing ionic adsorption, incomplete coverage or subsequent hydrolysis can re-expose active sites, and the resulting increase in hydrophobicity may exacerbate losses of lipophilic analytes unless mitigated by organic modifiers in the sample solvent.
Polymeric Surfaces
Polypropylene and polyethylene, common in pipette tips, vials, caps, and tubing, interact with analytes primarily through hydrophobic interactions, wetting behavior, and nonspecific partitioning onto or into the polymer surface. These risks are most apparent at low concentration, where small absolute losses become large relative recovery errors. Peptides, proteins, hydrophobic small molecules, amphiphilic analytes, and some oligonucleotide formulations can be especially sensitive to repeated contact with polymeric consumables. Low-binding or surface-engineered polymer consumables can reduce these losses, but they should not be treated as universally inert. Extractables, such as antioxidants, slip agents, plasticizers, and silicone-related contaminants, can introduce background signals and matrix effects, particularly in LC–MS workflows.
Metallic and Metal-Oxide Surfaces
Stainless steel (316L) passivates with a thin chromium- and iron-rich oxide/hydroxide layer. Depending on pH, ionic strength, surface treatment, and mobile-phase history, this layer can present charged and Lewis-acidic sites that interact with anionic or Lewis-basic analytes, including carboxylates, phosphates, sulfates, and oligonucleotides [2], [3]. Under-coordinated transition-metal centers can promote coordination interactions with Lewis-basic functional groups. At the same time, trace metal release can contribute to oxidation or detectable adduct formation in sensitive LC–MS workflows. Titanium and zirconia present related oxide chemistries; zirconia’s Lewis-acidic character has also been deliberately exploited for chromatographic selectivity [15]. Surface area scaling is critical: sintered-metal frits can provide far more active surface area than smooth tubing, making them potential dominant loss sites for acidic or phosphorylated analytes. Inert or hybrid-coated metal components reduce this risk by masking oxide sites while retaining the mechanical strength and pressure tolerance of metal hardware.
Table 1. Summary of principal surface–analyte interactions across materials commonly encountered in chromatographic workflows. Losses are illustrative and concentration-, pH-, and matrix-dependent.
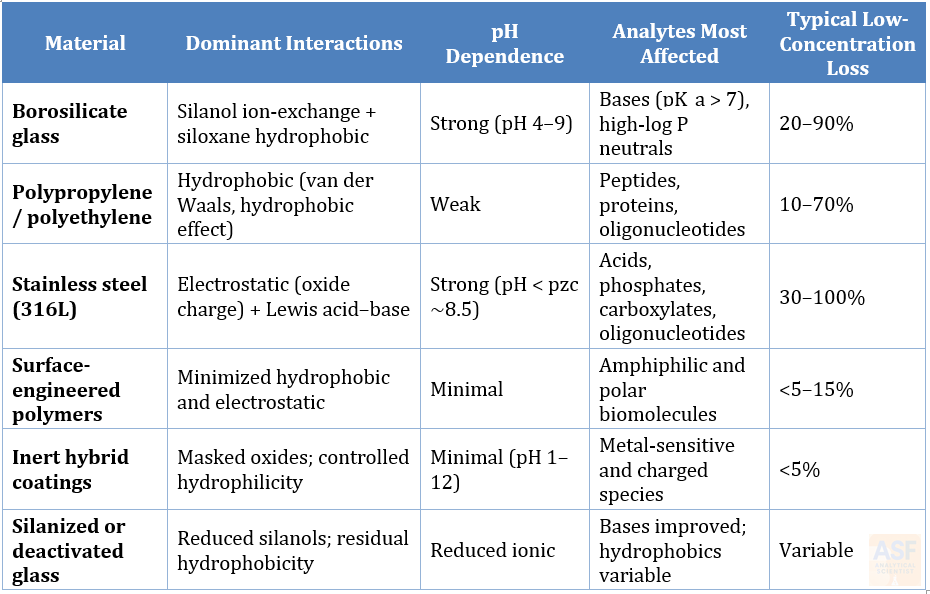
These surface classes are not equally important for every analyte. The practical question is therefore not whether adsorption is possible, but whether the analyte, concentration, matrix, and separation mode make adsorption analytically consequential.
Risk-Based Classification of Analytes by Surface Interaction Potential
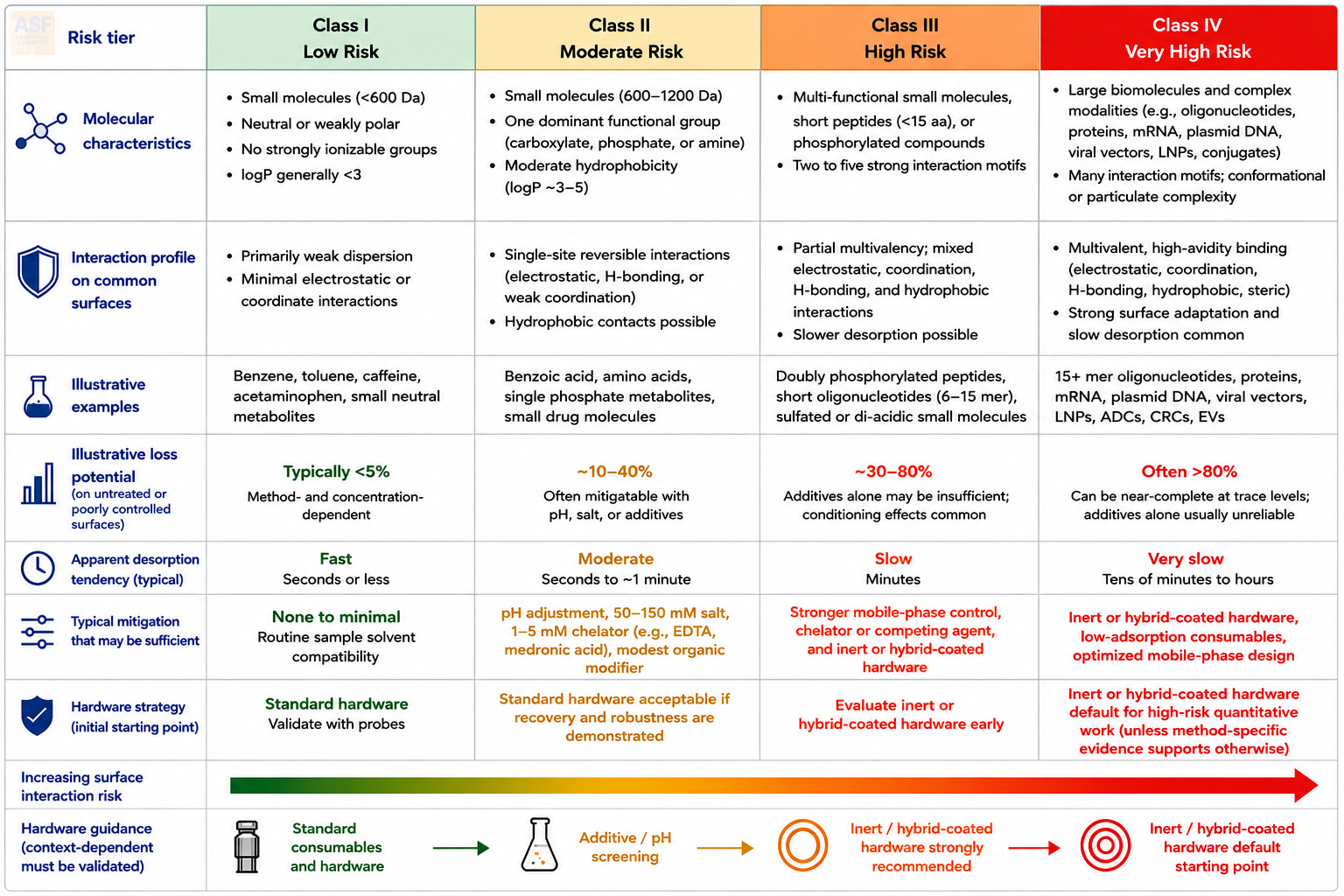
A practical way to operationalize surface chemistry is to classify analytes by their probability of analytically significant surface loss, tailing, carryover, or recovery bias. The proposed Class I–IV framework integrates molecular size, charge density, number of Lewis-basic or hydrophobic motifs, conformational flexibility, and expected working concentration. It is not a universal ranking of molecular importance. It is a method-development tool for deciding when standard hardware is adequate, when additive screening is reasonable, and when inert or hybrid-coated surfaces should be built into the method from the beginning.
Class I – Low Risk
Neutral or weakly polar small molecules (<600 Da) with no strongly ionizable groups and low to moderate logP (<3). These analytes usually interact through weak dispersion forces. Surface losses are often minor under conventional LC conditions, provided the working concentration is not extremely low, and the sample solvent is compatible with the workflow. Standard polypropylene or glass consumables are often adequate after routine recovery and carryover checks.Class II – Moderate Risk
Small molecules (600–1200 Da) bearing one dominant functional group (single carboxylate, phosphate, or amine) or moderate hydrophobicity (logP 3–5). Binding is largely reversible and follows simple Langmuir kinetics. Losses on untreated surfaces can become significant, especially at low concentration, but they are often reduced by pH adjustment, competing ions, or low concentrations of chelators such as ethylenediaminetetraacetic acid (EDTA) or medronic acid. Most small-molecule pharmaceuticals and metabolites fall into this class. Conventional hardware may be sufficient when recovery, carryover, and robustness are demonstrated.Class III – High Risk
Multi-functional small molecules, short peptides (<15 residues), or phosphorylated compounds with 2–5 strong interaction motifs. These analytes exhibit partial multivalency and can show slower desorption kinetics than Class I or II analytes. Recovery losses, carryover, and conditioning effects may become pronounced on stainless steel, untreated glass, or high-surface-area frits. Buffer optimization alone may be insufficient. Inert or hybrid-coated hardware should be evaluated early, especially for trace-level or regulated work. Representative probes include doubly phosphorylated peptides and short oligonucleotides.Class IV – Very High Risk
Large biomolecules and complex modalities, including longer oligonucleotides, proteins, mRNA, plasmid DNA, viral vectors, lipid nanoparticles, and conjugates, can present many interaction motifs and significant conformational or particulate complexity. These analytes can engage in cooperative, high-avidity binding that combines electrostatic, coordination, hydrogen bonding, hydrophobic, and steric interactions. On untreated or poorly controlled surfaces, recovery loss, carryover, adsorption hysteresis, and conditioning effects can become severe. For reproducible quantitation at relevant concentrations, inert or hybrid-coated flow paths and low-adsorption consumables should be considered default starting points unless method-specific evidence supports a simpler configuration.
The value of the framework is practical. Class I and II methods may be developed on standard hardware if recovery, precision, carryover, and robustness are demonstrated. Class III and IV methods should include early comparisons of standard and inert flow paths, recovery mapping across consumables, and system-suitability probes sensitive to surface activity. The decision should be evidence-based and documented in the analytical target profile and lifecycle control strategy.
Surface Chemistry Challenges by Separation Mode
Different chromatographic modes present distinct surface interaction risks because the mobile-phase composition, pH, ionic strength, and stationary-phase chemistry change the dominant forces at play. The table below summarizes the primary surface chemistry issues for each major mode, the most vulnerable analyte classes, and recommended hardware strategies.
Table 2. Surface Chemistry Issues by Separation Mode
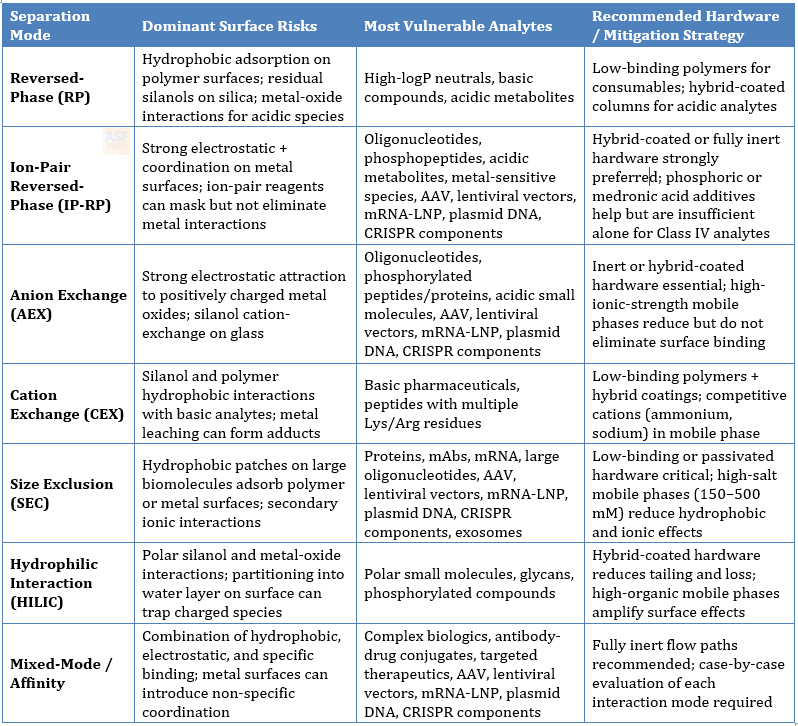
Table 2 emphasizes that surface risk changes with separation mode. Mobile-phase pH, salt concentration, organic content, ion-pairing reagent, detector choice, and analyte class all influence which surfaces become critical. Hardware selection should therefore be made alongside method design, not deferred until recovery, carryover, or transfer problems appear.
Sample Preparation and Storage: The First Critical Interfaces
The initial surfaces an analyte encounters are those of pipette tips, syringes, filters, and storage vials. Standard polypropylene tips can cause substantial losses of peptides, proteins, or oligonucleotides at sub-microgram-per-milliliter concentrations, especially after repeated aspirate–dispense cycles. Low-binding tips engineered to reduce adsorption can improve recovery for susceptible analytes. Filtration membranes introduce additional variables: nylon retains basic and aromatic compounds via hydrogen bonding and π–π interactions [5], while polytetrafluoroethylene (PTFE) is often suitable for organic-rich workflows but can retain highly hydrophobic analytes. Certified low-extractable filters are essential for liquid chromatography–mass spectrometry (LC–MS) workflows.
Vial walls present the same glass or polymer chemistry discussed above. For basic analytes, acidification (0.1–1% formic or phosphoric acid) suppresses silanol ionization, while addition of 50–150 mM ammonium or sodium salts competes for exchange sites. Organic modifiers (10–30% acetonitrile or methanol) and trace non-ionic surfactants further suppress hydrophobic contributions. Plastic vials eliminate silanol issues but rely solely on purely hydrophobic adsorption; surface-oxidized or plasma-treated polymers offer measurable improvements in recovery for polar biomolecules. Caps and septa contribute additional risks through leaching of cyclic siloxanes or permeation of volatiles; pre-slit, low-bleed septa are preferred for trace work.
Fluidic Pathway: Tubing, Fittings, Valves, Injectors, and Frits
Stainless-steel tubing and fittings contribute substantial surface area and are primary vectors for metal-ion leaching and nonspecific adsorption of acidic analytes. Passivation with nitric acid or chelator solutions provides only temporary relief. Inert polymer tubing, such as PEEK, can reduce metal-mediated interactions within its pressure and solvent-compatibility limits. Advanced inert fluidic pathways incorporating organic–inorganic hybrid coatings on metal components can combine high-pressure capability with reduced oxide-mediated adsorption, thereby reducing the need for frequent reconditioning.
Injection valves, needles, and sample loops experience repeated sample contact; stainless-steel needles can adsorb or catalyze degradation of thiols, catechols, and certain antibiotics. PEEK or passivated alternatives are preferred. The highest-surface-area components (column inlet and outlet frits and inline filters) dominate losses for acidic species. Fresh stainless-steel or titanium frits can produce severe adsorption of phosphorylated compounds and oligonucleotides under some mobile-phase conditions; successive injections gradually saturate sites, producing the familiar “conditioning” effect. Strong Lewis-base additives (phosphoric, citric, or etidronic acid) or chelators provide temporary passivation, but gradual desorption by the mobile phase necessitates periodic re-treatment. Hybrid surface modifications on frits and hardware can reduce the need for repeated passivation, delivering consistent recoveries from the first injection.
Column Hardware, Stationary-Phase Supports, and Detector Interfaces
Column tubes, frits, and particle substrates introduce additional uncontrolled surfaces. While bonded phases are engineered for selectivity, residual silanols on silica or Lewis-acid sites on alternative metal-oxide particles can cause tailing or irreversible loss of sensitive analytes. Inert or hybrid-coated column hardware mitigates these effects without sacrificing pressure performance or pH stability.
In optical detectors, flow-cell windows and wetted surfaces can contribute to fouling, adsorption, or baseline instability during extended use, particularly for proteins, lipid-containing samples, and particulate modalities. Flow-cell materials and cleaning protocols should therefore be considered part of the method control strategy when surface-sensitive analytes are analyzed. Electrospray ionization interfaces introduce additional surface and gas-phase complications: stainless-steel spray needles can adsorb analytes or introduce metal adducts, while the high-voltage region and heated transfer lines may catalyze oxidation of labile species. Inert or coated needles and low-adsorption flow paths from vial to source are increasingly used for trace bioanalysis and omics applications.
Surface and interfacial characterization methods help explain why empirical conditioning can be difficult to reproduce. QCM-D can monitor adsorption and viscoelastic changes at interfaces; AFM can probe surface morphology and local interaction forces; XPS and ToF-SIMS can characterize surface composition, oxidation state, and adsorbed residues. Together, these methods show that adsorption is not simply a bulk-solution problem. It depends on local charge, oxide chemistry, roughness, wetting, adsorbed contaminants, and surface history. For routine analytical laboratories, the practical implication is clear: conditioning protocols should be documented, challenged with relevant probes, and replaced by inert hardware when recovery or transfer risk is unacceptable.
For robust small-molecule assays at moderate concentrations, legacy stainless-steel systems, combined with documented passivation, additive screening, and appropriate system-suitability probes, may be scientifically adequate. For oligonucleotides, phosphopeptides, mRNA-related analytes, metal-sensitive metabolites, and trace-level biomolecule assays, however, the cost of adsorption-driven bias, failed transfer, or repeated reconditioning can outweigh the incremental cost of inert flow paths. The decision should be based on analyte risk, concentration, matrix, separation mode, detector requirements, and the consequences of recovery bias.
Advanced Therapeutic Modalities: Where Surface Risk Becomes System-Level Risk
Surface interactions become more consequential in advanced therapeutic modalities because the analytes are larger, more heterogeneous, more labile, and often more structurally complex than conventional small molecules. AAVs, lentiviral vectors, extracellular vesicles, mRNA-LNPs, plasmid DNA, CRISPR ribonucleoproteins, and oligonucleotide conjugates all combine large molecular size with charge heterogeneity, hydrophobic domains, conformational flexibility, or particulate architecture. Their interaction with glass, polymers, stainless steel, frits, filters, and detector interfaces is therefore rarely governed by a single mechanism. Adsorption can alter recovery, apparent purity, aggregate profiles, empty-to-full ratios, or payload measurements. For these modalities, surface interactions that may be tolerable in traditional small-molecule analysis become sources of method bias, transfer variability, or misleading product-quality readouts. Inert hardware and low-adsorption consumables should be built into method development from the beginning rather than introduced after transfer failure.
Mitigation Strategies Grounded in Surface Science
Mitigation should be proportional to the analyte risk and the method's consequences. For low-risk analytes, simple controls may be sufficient. For high-risk analytes, especially oligonucleotides, phosphopeptides, proteins, mRNA-related analytes, and gene-therapy materials, mitigation must combine material selection, mobile-phase design, hardware inertness, diagnostic recovery experiments, and lifecycle controls. The goal is not to make every method more complex. The goal is to match the control strategy to the probability and consequence of surface-driven bias.
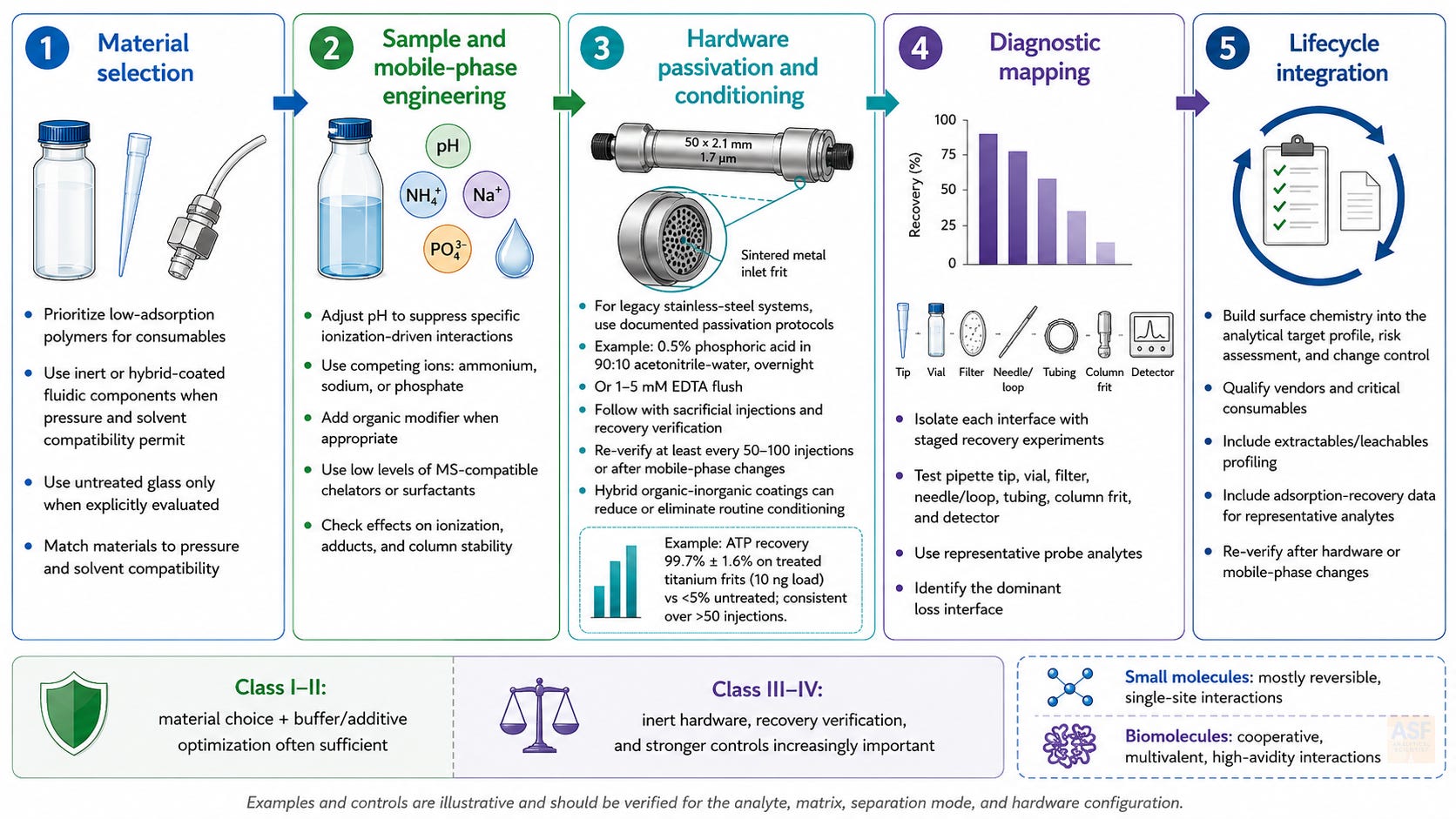
Material selection. Use low-adsorption polymer consumables for sample handling when recovery risk is high. Use inert or hybrid-coated flow paths when quantifying metal-sensitive, highly charged, phosphorylated, or multivalent analytes. Untreated glass or stainless steel should be used for high-risk analytes only when recovery and carryover have been demonstrated for the intended analyte and concentration range.
Sample and mobile-phase design. Use pH, ionic strength, organic modifiers, competing ions, chelators, or surfactants to suppress specific interactions, provided they are compatible with the separation and detector. These additives must be evaluated for secondary effects, including ion suppression, adduct formation, column lifetime, microbial growth, and method transferability.
Passivation and conditioning. For legacy stainless-steel systems, passivation and sacrificial injections can reduce active sites, but their effect is conditional and time-dependent. Any conditioning protocol should specify the reagent, concentration, contact time, rinse procedure, requalification criteria, and the trigger for repeat treatment. For high-risk Class III and IV analytes, inert hardware is often a more reproducible control than repeated empirical conditioning.
Diagnostic mapping. Perform staged recovery experiments that isolate each interface: sample container, pipette tip, filter, needle, loop, tubing, valve, frit, column hardware, and detector flow path. Use representative probes in addition to the final product, especially when early screening material is limited.
Lifecycle integration. Include surface chemistry in the analytical target profile, risk assessment, robustness plan, transfer protocol, system suitability, and change control. Critical consumables should be qualified not only for dimensions and cleanliness, but also for adsorption recovery and extractables or leachables risk when relevant.
Analytical Science Considerations: Implications for Method Lifecycle and Data Integrity
Surface chemistry should be controlled across the analytical procedure lifecycle. Under ICH Q14 and USP <1220>, method development should define the intended purpose of the procedure, identify variables that affect performance, and establish a control strategy. For analytes with significant surface-interaction risk, that control strategy should include the sample container, preparation workflow, filters, fluidic pathway, column hardware, detector interface, and any conditioning or passivation procedure.
During development, recovery should be mapped by spiking before and after discrete handling steps. During validation, robustness studies should challenge representative consumables, hardware states, and relevant hold times. During transfer, the receiving laboratory should verify not only the column and mobile phase, but also vial type, filter type, needle material, tubing, frits, and system history. During routine use, peak area, tailing factor, carryover, adduct formation, and probe analyte recovery can be trended to detect surface changes before they affect reportable results.
This approach turns surface chemistry from an undocumented variable into a lifecycle-controlled parameter. This control is most important when analytical results sit near decision boundaries: impurity limits, potency-related assays, empty-to-full ratio assessments, trace-level quantification, degradation monitoring, and batch release.
Conclusion
Surface chemistry is no longer a peripheral concern in liquid chromatography. It affects recovery, sensitivity, carryover, peak shape, adduct formation, transferability, and the credibility of data used for development and release decisions. As analytical targets shift from conventional small molecules toward oligonucleotides, mRNA-LNPs, viral vectors, plasmid DNA, proteins, conjugates, and other advanced modalities, the surfaces surrounding the column can become as consequential as the column itself.
The proposed Class I–IV framework provides a practical way to match analytical controls to analyte risk. Low-risk methods may require only conventional robustness checks. High-risk methods should include low-adsorption consumables, inert or hybrid-coated flow paths, staged recovery studies, surface-activity-sensitive system suitability, and lifecycle change control. The point is not to mandate expensive hardware for every assay. The point is to prevent surface-driven bias from being mistaken for chemistry, degradation, impurity, poor recovery, or biological variability.
Chromatographic data are generated by the entire workflow, not just the stationary phase. Every vial, tip, filter, needle, loop, frit, tube, column wall, and detector surface can either preserve or distort the analytical result. Treating those interfaces as controlled method variables is now essential for robust LC in oligonucleotide, mRNA, gene-therapy, and complex biologics development.
References
[1] G. J. Guimaraes and M. G. Bartlett, “Managing nonspecific adsorption to liquid chromatography hardware: A review,” Anal. Chim. Acta, vol. 1250, Art. no. 340994, 2023, doi: 10.1016/j.aca.2023.340994.
[2] M. Gilar, M. DeLano, and F. Gritti, “Mitigation of analyte loss on metal surfaces in liquid chromatography,” J. Chromatogr. A, vol. 1650, Art. no. 462247, 2021, doi: 10.1016/j.chroma.2021.462247.
[3] T. Nagayasu et al., “Effects of carboxyl groups on the adsorption behavior of low-molecular-weight substances on a stainless steel surface,” J. Colloid Interface Sci., vol. 279, no. 2, pp. 296–306, 2004, doi: 10.1016/j.jcis.2004.06.081.
[4] H. Lardeux et al., “The impact of low adsorption surfaces for the analysis of DNA and RNA oligonucleotides,” J. Chromatogr. A, vol. 1677, Art. no. 463324, 2022, doi: 10.1016/j.chroma.2022.463324.
[5] K. Maes et al., “Strategies to reduce aspecific adsorption of peptides and proteins in liquid chromatography–mass spectrometry based bioanalyses: An overview,” J. Chromatogr. A, vol. 1358, pp. 1–13, 2014, doi: 10.1016/j.chroma.2014.06.072.
[6] International Council for Harmonisation, “ICH Q2(R2): Validation of Analytical Procedures,” final version, adopted Nov. 1, 2023. [Online]. Available: https://www.ich.org/page/quality-guidelines
[7] International Council for Harmonisation, “ICH Q14: Analytical Procedure Development,” final version, adopted Nov. 1, 2023. [Online]. Available: https://www.ich.org/page/quality-guidelines
[8] United States Pharmacopeia and National Formulary, “USP–NF General Chapter <1220> Analytical Procedure Life Cycle,” current official text.
[9] D. R. Stoll, “Strongly adsorbing analytes: What, why, and how to fix it,” LCGC North America, vol. 41, no. 7, pp. 242–244, 2023, doi: 10.56530/lcgc.na.vr7280f7.
[10] J. J. Hsiao, T.-W. Chu, O. G. Potter, G. O. Staples, and D. R. Stoll, “Troubleshooting LC separations of biomolecules, part 2: Passivation and mobile-phase additives,” LCGC North America, vol. 38, no. 8, pp. 431–434, 2020.
[11] M. Galmiche et al., “Substantial benefits of an inert biphenyl column for the analysis of steroids and their phase II metabolites in biological samples,” J. Sep. Sci., vol. 47, no. 16, Art. no. e2400436, 2024, doi: 10.1002/jssc.202400436.
[12] D. Kumar, M. Sharma, and N. Trivedi, “A roadmap guide on bioanalysis challenges and practical solutions for accurate quantification of oligonucleotide-based novel therapeutic modalities using LC-MS,” J. Chromatogr. B, vol. 1270, Art. no. 124900, 2026, doi: 10.1016/j.jchromb.2025.124900.
[13] D. S. Bell, “Evolutions in particle, surface chemistry, and hardware designs: New liquid chromatography (LC) columns and accessories for 2024,” LCGC International, vol. 1, no. 5, pp. 14–22, 2024, doi: 10.56530/lcgc.int.fp6887j6.
[14] A. Murisier, S. Fekete, D. Guillarme, and V. D’Atri, “The importance of being metal-free: The critical choice of column hardware for size exclusion chromatography coupled to high resolution mass spectrometry,” Anal. Chim. Acta, vol. 1183, Art. no. 338987, 2021, doi: 10.1016/j.aca.2021.338987.
[15] J. A. Blackwell and P. W. Carr, “Development of an eluotropic series for the chromatography of Lewis bases on zirconium oxide,” Anal. Chem., vol. 64, no. 8, pp. 863–873, 1992, doi: 10.1021/ac00032a008.
[16] M. DeLano et al., “Using hybrid organic–inorganic surface technology to mitigate analyte interactions with metal surfaces in UHPLC,” Anal. Chem., vol. 93, no. 14, pp. 5773–5781, 2021, doi: 10.1021/acs.analchem.0c05203.
[17] L. R. Snyder, J. J. Kirkland, and J. W. Dolan, Introduction to Modern Liquid Chromatography, 3rd ed. Hoboken, NJ, USA: Wiley, 2010.
[18] R. P. W. Scott, Silica Gel and Bonded Phases: Their Production, Properties and Use in LC. Chichester, U.K.: Wiley, 1993.