
Restoring Hearing: The First Approved Gene Therapy for Genetic Hearing Loss
Dual-AAV therapy for OTOF-related deafness receives accelerated approval, but confirmatory data on durability and functional outcomes remain pending through 2030


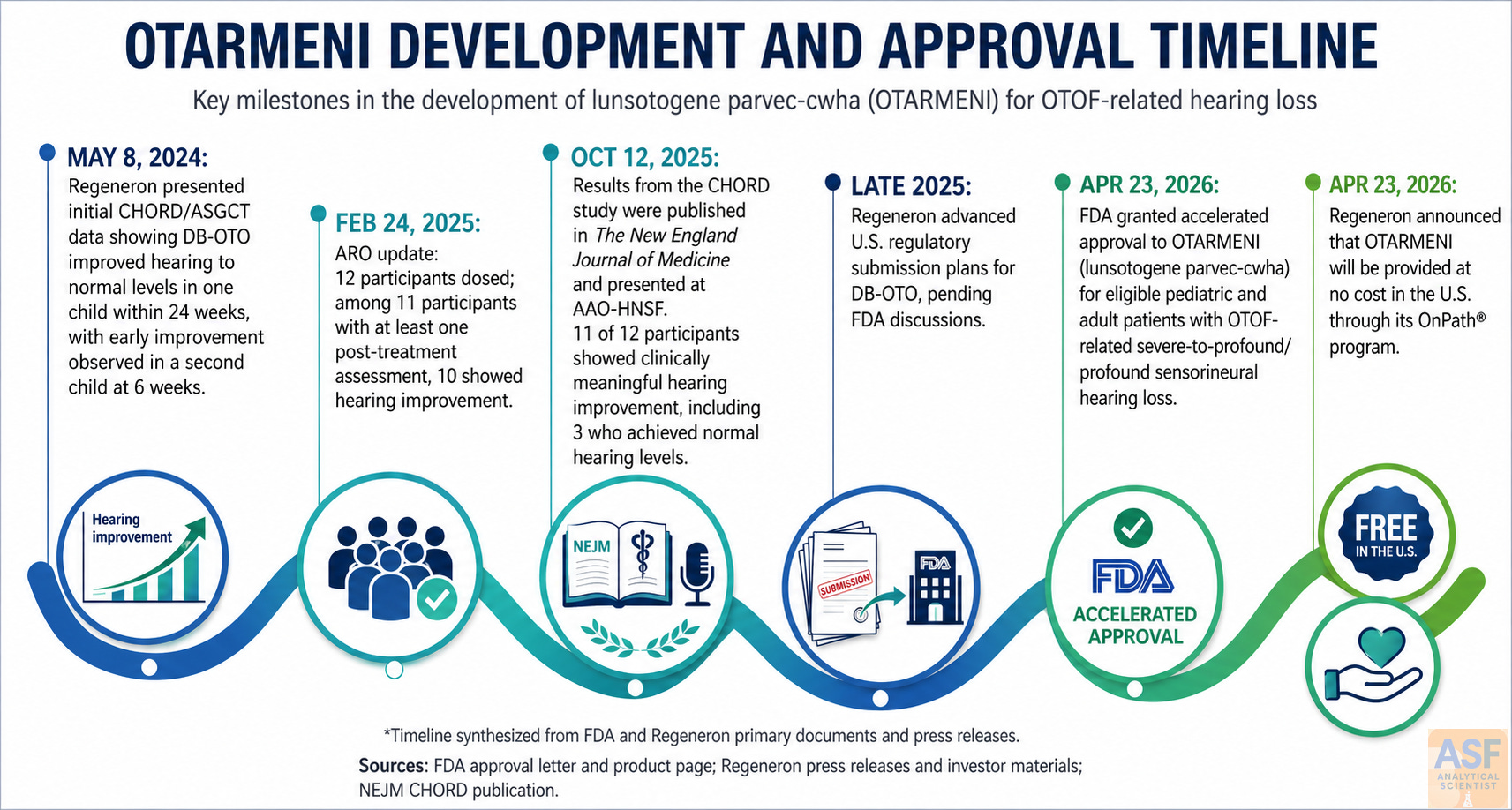
On 23 April 2026, the U.S. Food and Drug Administration (FDA) granted accelerated approval to lunsotogene parvec-cwha (Otarmeni), the first gene therapy for genetic hearing loss and the first Dual adeno-associated virus (AAV) therapy for OTOF-related deafness receives accelerated approval product approved for any indication.1,2 The one-time intracochlear therapy targets biallelic OTOF variants that cause profound congenital deafness by restoring otoferlin expression in inner hair cells. Regeneron simultaneously announced no-cost access for eligible U.S. patients, although surgical and facility costs remain the patient’s responsibility.3,12
The approval rests on persuasive but provisional data from an ongoing open-label, single-arm trial. Hearing thresholds improved substantially in most treated children, yet long-term durability, speech and language development, and quality-of-life benefits are unproven. The FDA, therefore, required extensive confirmatory and post-marketing studies extending through 2040. This represents a genuine therapeutic advance for a previously untreatable genetic deafness, delivered with appropriate regulatory caution.
Approval Details and Access
The indication covers pediatric and adult patients with severe-to-profound or profound sensorineural hearing loss (any frequency >90 dB HL) due to molecularly confirmed biallelic OTOF variants, preserved outer hair cell function, and no prior cochlear implant in the treated ear.1 Treatment is contraindicated only when preoperative imaging shows anatomy incompatible with safe intracochlear access. The label lists no contraindications, carries no boxed warning, and imposes no Risk Evaluation and Mitigation Strategy (REMS).1,2
Warnings focus on standard risks of mastoidectomy and round-window infusion: vertigo, tinnitus, cerebrospinal fluid leak, facial paresis, taste changes, meningitis, and wound complications. The The Biologics License Application (BLA) was reviewed in 61 days under the National Priority Voucher pilot, making Otarmeni the first dual-AAV gene therapy and the first product approved via that pathway.11,14
Why OTOF Disease Is Tractable
Genetic causes account for roughly 80% of prelingual hearing loss, and U.S. programs screen more than 98% of newborns.5 Most genetic deafness subtypes are mechanistically complex, but OTOF-related disease is an auditory synaptopathy: inner hair cells detect sound (preserved otoacoustic emissions) yet fail to transmit signals because otoferlin is absent or nonfunctional.6 This explains the requirement for preserved outer hair cell function and why otoacoustic-emissions-only screening misses these infants.
Pathogenic OTOF variants cause up to 8% of congenital nonsyndromic hearing loss and 41–91% of auditory neuropathy spectrum disorder cases tested.6 A systematic review of 422 individuals confirms phenotypic heterogeneity, including rare progressive forms.7 The label is therefore appropriately narrow.
Therapeutic Design and Administration
Otarmeni comprises two complementary AAV1 vectors that together encode full-length human otoferlin isoform e driven by a hair-cell-specific Myo15 promoter.1,8 The dual-vector approach overcomes the size limit of single AAV particles for the approximately 6 kb OTOF coding sequence. Preclinical work in Otof-deficient mice demonstrated sustained functional hearing recovery.8,9
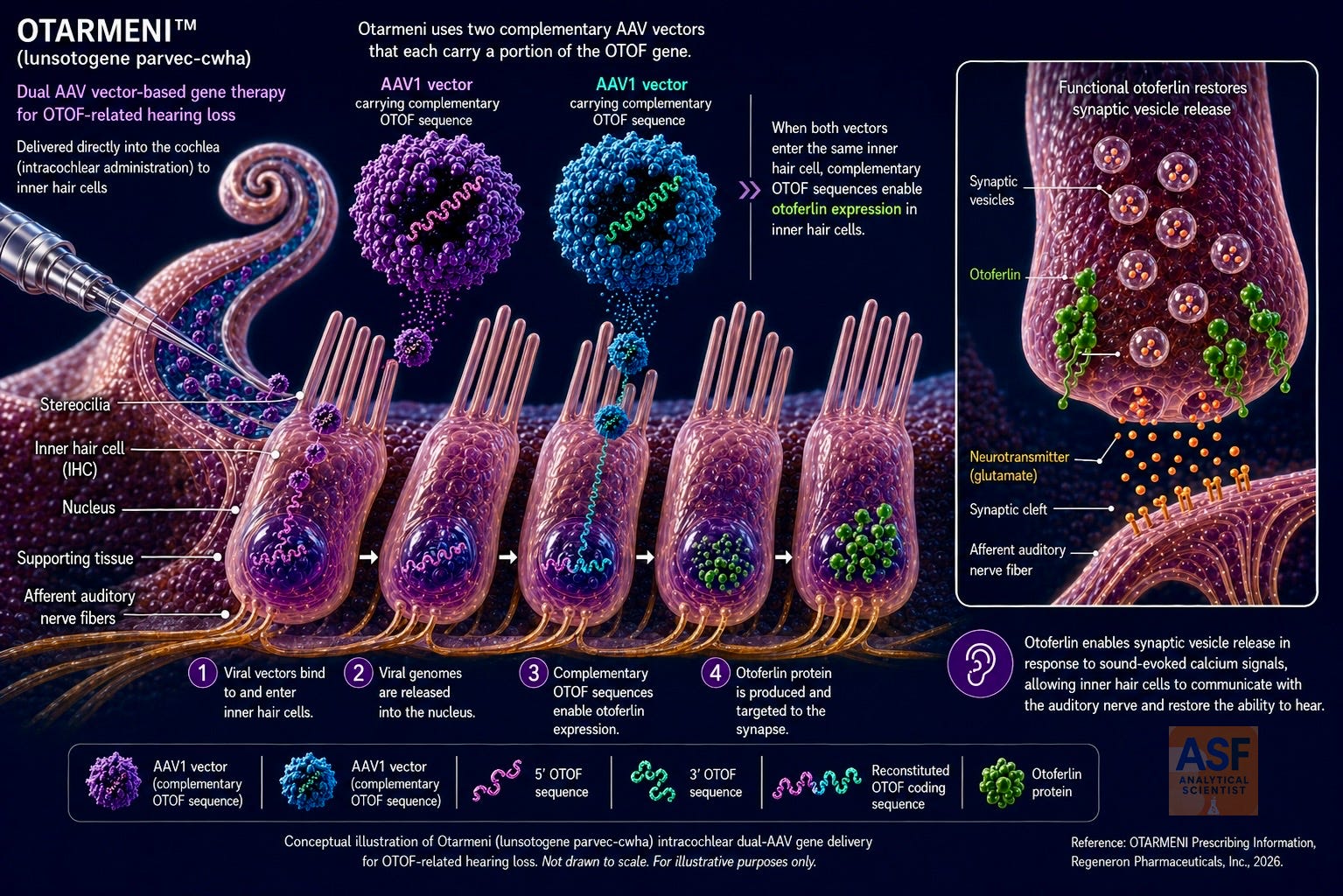
The dose is 7.2 × 10¹² vector genomes per ear in 0.24 mL, given once by intracochlear infusion after mastoidectomy and round-window access. Infusion lasts 16 minutes at 0.9 mL/h followed by a 5-minute dwell. A dedicated kit supplies the catheter and syringes; the pump is user-supplied. Perioperative care includes a 4-week corticosteroid taper (prednisone 1 mg/kg/day, maximum 60 mg), prophylactic antibiotics, and carefully timed vaccinations. Body-fluid precautions apply for up to 12 weeks post-dose.1
CHORD Trial Design and Datasets
Approval is based on DB-OTO-001 (CHORD; NCT05788536), an ongoing first-in-human, open-label, single-arm study.1,4 Public evidence consists of sequential snapshots of the same trial. The New England Journal of Medicine reported a 12-patient registrational analysis4,10; the FDA label uses a 24-patient safety population (20 evaluable at Week 24, 12 at Week 48).1 These are not inconsistent; the study matured between publication and approval.
Table 1. Evolution of clinical datasets from DB-OTO-001 (CHORD). All data come from the same ongoing open-label, single-arm trial*. The approval incorporates later, larger cuts than the initial publication. Bilateral efficacy uses the better-treated ear.1,3,4,10
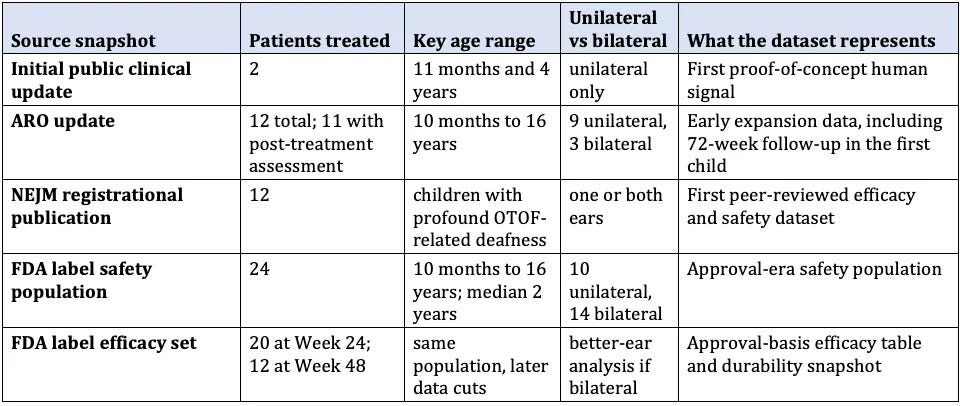
*Eligibility required age under 18 at dosing (label extends to adults), biallelic pathogenic or likely pathogenic OTOF variants, profound sensorineural hearing loss, and preserved outer hair cell function by otoacoustic emissions or cochlear microphonic. Exclusions included prior gene therapy, incompatible anatomy, cochlear implant in the ear to be treated, malignancy, and alternative auditory neuropathy etiologies.1,4
Efficacy Outcomes
Table 2. Hearing sensitivity outcomes in CHORD. PTA = pure-tone average (dB HL); ABR = auditory brainstem response (dB nHL). The NEJM dataset is smaller and earlier; FDA data reflect additional patients and later follow-up.1, 4
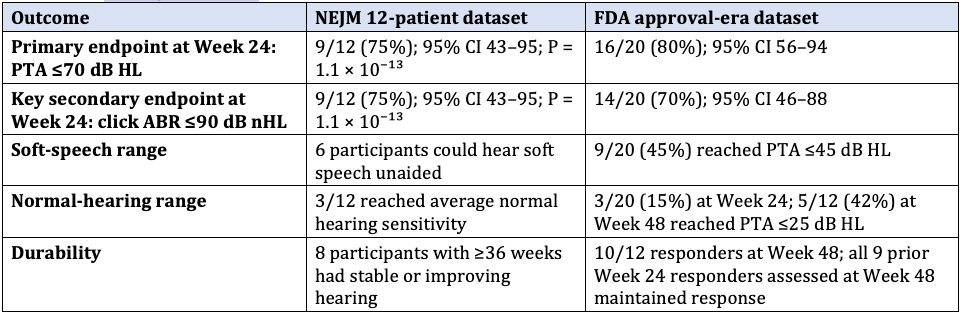
In the 12-patient publication, six children heard soft speech without aids, and three reached normal sensitivity. In the larger FDA set, 45% reached the soft conversational speech range, and 15% reached the normal range at Week 24; at Week 48, 42% reached the normal range.1,4 These gains are clinically meaningful, yet the open-label design and absence of a control arm limit causal attribution. Speech-in-noise perception and language acquisition data are sparse and constitute core endpoints of the required confirmatory program.
Safety
Table 3. Most frequent adverse events in the 24-patient safety population. Rates are consistent with inner-ear surgery under general anesthesia. No events led to discontinuation. Labeled risks also include facial paresis, cerebrospinal fluid leak, and meningitis (rare).1
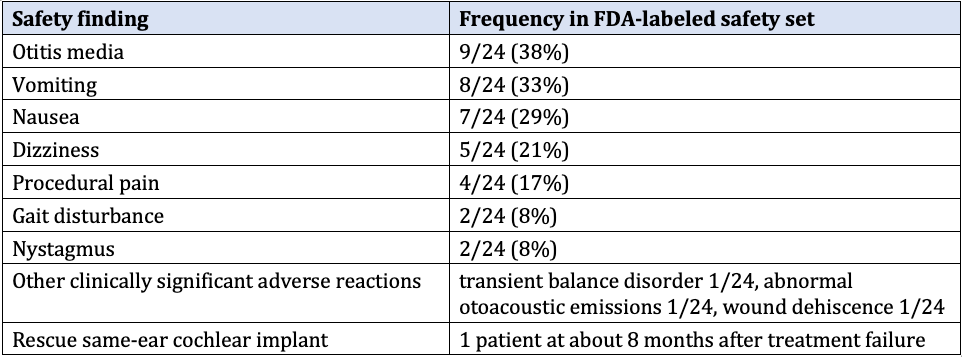
Immunogenicity was common (anti-AAV1 neutralizing antibodies in all 15 patients tested through Week 12; anti-OTOF antibodies in 7 of 15), but it did not appear to affect safety or efficacy.1 Vector shedding was transient (undetectable in plasma, urine, and saliva by Week 2; in stool by Week 12). Biodistribution in animals was largely cochlear, with minimal non-otic spread and no reproductive-tissue expression of human OTOF messenger RNA (mRNA).
Regulatory Commitments and Timeline
Accelerated approval was based on a surrogate endpoint (hearing sensitivity) reasonably likely to predict clinical benefit. The FDA therefore mandated a confirmatory program: at least 30 treated pediatric patients followed for 104 weeks plus at least 5 newly treated patients aged 16 years or older, compared with untreated controls. Endpoints include PTA, age-appropriate speech and language measures, ABR, global impression scales, and quality-of-life instruments. Protocol due 30 December 2026; completion by 30 April 2030; final report 30 August 2030.11 A separate 10-year post-marketing safety study will enroll at least 50 patients with malignancy surveillance; interim reports due in 2029 and 2032, final report in 2040.11
Analytical Science Considerations
The approval has significant implications for analytical scientists working on complex gene therapies. Otarmeni demonstrates both the current capabilities and the future needs of analytical science in this field.
The dual-AAV1 platform, which needs to deliver the large OTOF coding sequence, faces specific challenges in maintaining vector genome integrity, improving recombination efficiency between the 5′ and 3′ vectors, and accurately measuring full versus partial or empty capsids. Regulatory requirements for AAV products now call for comprehensive analytical methods that include high-resolution size-exclusion chromatography combined with multi-angle light scattering (SEC-MALS), analytical ultracentrifugation (AUC) for assessing the ratio of empty-to-full capsids and detecting aggregates, anion-exchange chromatography, and highly sensitive tests for residual host-cell DNA and protein. Achieving an 80% primary-endpoint response rate in the key dataset shows that these approaches, when used carefully, were enough to support accelerated approval.
The hair-cell-specific Myo15 promoter adds extra requirements: sequence confirmation must be combined with functional potency tests that show a correlation to in vivo inner-hair-cell expression. As dual-AAV and other large-gene therapies grow in popularity, the field will need standardized reference materials and validated assays for split-vector recombination efficiency to ensure consistent lot release and comparability.
Transient shedding profiles seen in clinical studies highlight the need for highly sensitive, matrix-validated digital polymerase chain reaction (PCR) or quantitative PCR (qPCR) methods with clear detection limits. Comprehensive immunogenicity testing, covering both anti-capsid neutralizing antibodies and anti-transgene antibodies, remains crucial and should use tiered, multi-platform approaches.
Continued investment in higher-resolution techniques, especially advanced analytical methods like mass photometry and charge detection mass spectrometry (CDMS), along with integrated multi-attribute analytics, will be necessary to maintain product quality, consistency, and patient safety as the AAV pipeline grows. By directly measuring the mass-to-charge ratio of intact viral particles, CDMS allows for high-resolution differentiation of empty, partially filled, and fully packaged capsids and offers insights into genome packaging integrity. For dual-AAV products like lunsotogene parvec-cwha, this capability is especially useful due to the need for coordinated delivery of the two complementary vectors. Although not included in the current validated analytical suite supporting accelerated approval, CDMS and similar next-generation methods, combined with existing analytical tools, are expected to enhance future process understanding, comparability, long-term product consistency, quality, and patient safety as the AAV pipeline expands.
Conclusion
Otarmeni marks a milestone in medicine: the first approved gene therapy for genetic hearing loss and the first dual-AAV product to reach patients. The early hearing improvements observed in many treated children provide real hope to families who, until now, had no treatment options. While the lack of randomized controls and the need for confirmatory studies through 2030 mean the full clinical value is still being determined, this approval signals the start of a new era.
It is both a historic therapeutic milestone and a strong demonstration that accelerated pathways, when combined with rigorous science and long-term dedication, can deliver life-changing treatments. Families, clinicians, and regulators now stand together at the brink of a future where genetic hearing loss may no longer be permanent, and where this success opens the door for gene therapies targeting other types of deafness and beyond.
References
FDA. Package Insert: OTARMENI (lunsotogene parvec-cwha), revised April 2026. https://www.fda.gov/media/192098/download?attachment
FDA. OTARMENI product page. https://www.fda.gov/vaccines-blood-biologics/otarmeni
Regeneron Pharmaceuticals. Otarmeni approved by FDA as first and only gene therapy for genetic hearing loss; Regeneron to provide Otarmeni for free in the U.S. (23 April 2026). https://investor.regeneron.com/news-releases/news-release-details/otarmenitm-lunsotogene-parvec-cwha-approved-fda-first-and-only/
Valayannopoulos V, et al. DB-OTO gene therapy for inherited deafness. N Engl J Med. 2026;394(11):1074–1083. doi:10.1056/NEJMoa2400521
Shearer AE, et al. Genetic hearing loss overview. GeneReviews. Updated 2025. https://www.ncbi.nlm.nih.gov/books/NBK1434/
Azaiez H, et al. OTOF-related hearing loss. GeneReviews. Updated 2025. https://www.ncbi.nlm.nih.gov/books/NBK1251/
Ford CL, et al. The natural history, clinical outcomes, and genotype-phenotype relationship of otoferlin-related hearing loss: a systematic, quantitative literature review. Hum Genet. 2023;142(10):1429–1449. doi:10.1007/s00439-023-02595-5
Chung Y, et al. Functional, sustained recovery of hearing in Otoferlin-deficient mice using DB-OTO, a hair-cell-specific AAV-based gene therapy. Mol Ther Methods Clin Dev. 2025;33(4):101577. doi:10.1016/j.omtm.2025.101577
Sellon JB, et al. Recovery kinetics of dual AAV-mediated human otoferlin expression. Front Mol Neurosci. 2024;17:1376128. doi:10.3389/fnmol.2024.1376128
Regeneron Pharmaceuticals. DB-OTO results in the New England Journal of Medicine showcase dramatic and sustained improvements in hearing and speech perception in children with profound genetic hearing loss (12 October 2025). https://investor.regeneron.com/news-releases/news-release-details/db-oto-results-new-england-journal-medicine-showcase-dramatic/
FDA. Approval letter for lunsotogene parvec-cwha, BL 125874/0 (23 April 2026). https://www.fda.gov/media/192111/download?attachment
Regeneron Pharmaceuticals. Regeneron announces agreement with U.S. government to help lower drug costs for American patients and will provide innovative new gene therapy for free in the U.S. (23 April 2026). https://investor.regeneron.com/news-releases/news-release-details/regeneron-announces-agreement-us-government-help-lower-drug/
Regeneron Pharmaceuticals. Latest DB-OTO results demonstrate clinically meaningful hearing improvements in nearly all children with profound genetic hearing loss in CHORD trial (24 February 2025). https://investor.regeneron.com/news-releases/news-release-details/latest-db-oto-results-demonstrate-clinically-meaningful-hearing/
FDA. FDA approves first-ever gene therapy for treatment of genetic hearing loss under National Priority Voucher program (23 April 2026). https://www.fda.gov/news-events/press-announcements/fda-approves-first-ever-gene-therapy-treatment-genetic-hearing-loss-under-national-priority-voucher